AMBER Tutorial: Molecular Dynamics of Na+/Cl- Association¶
by Karl Debiec and Daniel Hatfield
Updated with WESTPA version 1.0 beta and Amber 14
Overview¶
Requirements: ~1 hr wallclock time on an 8-core Intel Westmere node (one walker per core); ~1.2 GB disk space
In this introductory tutorial we will use the weighted ensemble strategy to simulate Na+/Cl- association in Generalized Born implicit solvent. The system consists of single Na+ and Cl- ions modeled with the Amber force field, using the distance between the two ions as the progress coordinate. Amber will be used to run the molecular dynamics, and familiarity with it is a prerequisite (see tutorials). Basic knowledge of python and bash scripting is also necessary.
The first step is to set up a directory containing the necessary Amber and
WESTPA files. A working example directory can be found at
westpa/lib/examples/nacl_amb.
Preparing the Amber files¶
| Input File | Description |
|---|---|
| nacl.prm | topology |
| nacl.crd | coordinates |
| md-genvel.in | configuration for initial segments |
| md-continue.in | configuration for continuations |
The above files are included in the amber_config subfolder of the example
directory, and should be familiar to Amber users. Note that two configuration
files are present.
md-genvel.in is used for initial segments, in which it is necessary to
generate initial velocities, and md-continue.in is used for
continuations, in which velocities are inherited from the parent segment.
md-continue.in¶
0.5 ps NVT production with Langevin thermostat and GB implicit solvent
################################### CONTROL ###################################
&cntrl
#################################### INPUT ####################################
ntx = 5, # Read input coordinates and velocities
irest = 1, # Restart calculation from input file
ig = RAND, # Use random seed from WESTPA
################################# INTEGRATOR ##################################
dt = 0.002, # Timestep (ps)
nstlim = 250, # Simulation duration (timesteps)
################################## ENSEMBLE ###################################
ntt = 3, # Langevin thermostat
temp0 = 300.0, # System temperature (K)
gamma_ln = 0.5, # Langevin collision frequency (1 / tau)
(ps-1)
########################### NONBONDED INTERACTIONS ############################
cut = 999.0, # Nonbonded cutoff (A)
rgbmax = 999.0, # Cutoff for effective Born radii
calculation (A)
############################## IMPLICIT SOLVENT ###############################
igb = 1, # Generalized Born implicit solvent
################################### OUTPUT ####################################
ntpr = 50, # Log output interval (timesteps)
ntxo = 2, # Output restart file in NetCDF binary format
ntwr = 250, # Restart file output interval (timesteps)
ioutfm = 1, # Output trajectory in NetCDF binary format
ntwx = 50, # Trajectory output interval (timesteps)
&end
Note that the comments in this input file must be removed prior to use with Amber, uncommented versions are provided in the example directory.
Several settings in this configuration file are important to WESTPA. First is the the segment duration, τ, which is equal to the timestep times the number of steps. The appropriate τ depends on the system and progress coordinate, and for this system we have chosen 0.5 ps. Second is the use of the stochastic Langevin thermostat; WESTPA requires a stochastic element such that trajectories branched from a common point diverge. In order to ensure that this occurs, it is also necessary to set the random seed to a different value for each segment. Setting the random seed based on the time, as is common for brute-force simulations, may cause problems as simulations started simultaneously from the same parent will not diverge.
The analogous md-genvel.in differs only in that it does not load
velocities from the input file, instead generating them from a Maxwellian
distribution.
Preparing the WESTPA files¶
| Input File | Description |
|---|---|
| env.sh | set environment variables |
| gen_istate.sh | generate initial states from basis states |
| get_pcoord.sh | calculate progress coordinate for initial states |
| west.cfg | WESTPA configuration |
| aux_functions.py | functions for loading auxilliary data |
| runseg.sh | segment implementation |
| post_iter.sh | post-segment cleanup |
| init.sh | initialize WESTPA |
| run.sh | run WESTPA |
| tar_segs.sh | tar segments |
The above files are listed roughly in the order in which it is appropriate to
configure them. gen_istate.sh, get_pcoord.sh, runseg.sh, and
post_iter.sh are located in the westpa_scripts subfolder.
Typically when setting up a WESTPA simulation, you will need to write these files yourself or modify the files provided in an example, such as this tutorial.
env.sh¶
This script sets environment variables that may be used across the simulation. These include the root directory for WESTPA, the root directory for the simulation, the name for the simulation, and the python executable to use. It also sets the executables for Amber; using an environment variable for this purpose makes it easier to transition code to different hardware or test different builds or flags of an MD code without editing multiple files.
Make sure that your $AMBERHOME environment variable is correctly set and
that $AMBERHOME/lib is in $LD_LIBRARY_PATH. This should be the case if
you have sourced amber.sh from your Amber installation directory.
gen_istates.sh¶
This script generates initial states (configurations) for the simulation from
the basis state stored in the bstates subfolder. Our system uses a single
basis state containing the two ions with a separation of 9.90 Å; this script
generates slight variations of this distance in order to obtain a greater
variety of starting configurations.
get_pcoord.sh¶
This script calculates the progress coordinate for each of the initial states.
Our progress coordinate is the distance between the Na+ and Cl- ions, which we calculate using AmberTools’ cpptraj. Note that this
script is used only during initial state generation; throughout the remainder of
the weighted ensemble simulation, runseg.sh calculates the progress
coordinate.
west.cfg¶
- This file defines the WESTPA configuration, including:
- the number of dimensions of the progress coordinate
- the number of data points per trajectory segment
- the binning scheme and number of walkers per bin
- the number of weighted ensemble iterations to run
- the locations of various scripts
For this system we use 22 bins as defined by Zwier, Kaus, and Chong, and 24 walkers per bin.
aux_functions.py¶
In addition to the progress coordinate, WESTPA includes the ability to calculate and store auxiliary data as the simulation is run. This is often easier than looping over iterations and segments afterwards. Since our system contains only two atoms, it is reasonable for us to store all coordinate information in the same hdf5 file as the progress coordinate. We will also store the log information including time, energy, and temperature.
This file contains Python functions for loading the coordinates and other
auxilliary information. The location (module and function name) of these
functions is specified to WESTPA in west.cfg.
runseg.sh¶
This script runs individual segments, calculates and outputs their progress
coordinates, and outputs auxiliary data. For each iteration and segment it
generates a folder, linking to the files necessary for running Amber.
For the Amber configuration file, it uses sed to input a random seed
generated by WESTPA. It then runs Amber, calculates and outputs the progress
coordinate and auxiliary data, and removes files that are no longer needed. As
in get_pcoord.sh, the progress coordinate is calculated using AmberTools’
cpptraj.
The auxiliary coordinate dataset is also prepared using cpptraj, which is
used to convert to trajectory to pdb format, which is processed using shell
commands and output to a temporary file, from which it is read by the
coord_loader function in aux_functions.py.
The auxiliary log data is similarly processed using shell commands and output
to a temporary file, from which it is further processed and stored by the
log_loader function in aux_functions.py.
post_iter.sh¶
This script cleans up after each iteration. WESTPA simulations can generate
large numbers of files, potentially conflicting with filesystem restrictions.
After each iteration, post_iter.sh packages the segment logs as a tar
file.
init.sh¶
This script initializes the WESTPA system. It removes files from previous runs
and uses gen_istates.sh and get_pcoord.sh to generate initial states. This
is also where the basis states and target states are defined. For this system
we define the bound target state as 1.8 Å separation, and the unbound target
state as 16.9 Å separation. Once walkers reach the bins containing these values
(i.e. our first and final bins), they are recycled. init.sh is also one of
two places where we specify the number of walkers per bin, as
--segs-per-state.
run.sh¶
This script is used to run WESTPA.
tar_segs.sh¶
This script is used to tar segments after the WESTPA simulation has been run,
in order to reduce the number of files produced. In order to allow extension of
the simulation, the last segment is not tarred. Typically, it is advisable not
to tar segments after each iteration (i.e. in post_iter.sh), while the main
WESTPA process is tarring, other cores are idle, potentially wasting CPU time.
Running the simulation¶
Make sure that the environment variable $WEST_ROOT is set and points your
WESTPA installation. To set this variable, enter
export WEST_ROOT=/path/to/your/westpa/installation at the terminal,
modifying the command with the appropriate path. You can check if
$WEST_ROOT is set by typing echo $WEST_ROOT at the terminal.
From the simulation root directory ($WEST_SIM_ROOT), the simulation may be
initialized using the command:
./init.sh
and run using the command:
./run.sh
init.sh and run.sh call w_init and w_run from WESTPA. By
default WESTPA will use as many cores as are available on the host machine. If
the simulation is run on a computing cluster, w_run may be executed from
a batch script. See the Running page for more information on how to
submit jobs to specific clusters.
Analyzing the data¶
Output¶
| Output File | Remarks |
|---|---|
| traj_segs | output from each iteration and segment |
| seg_logs | log files from each iteration and segment |
| west.h5 | WESTPA output in hdf5 database |
| west.log | WESTPA log file |
traj_segs¶
This folder stores the results of the WESTPA simulation, organized by iteration
and segment. This includes all files generated by runseg.sh, including
those generated by Amber.
For this system, the only files saved are seg.crd, seg.rst, and
seg.log corresponding to the coordinates, final restart, and log.
After the simulation has been run, tar_segs.sh may be used to reduce each
iteration to a single tar file.
seg_logs¶
This folder stores logs from each iteration and segment. After each weighted
ensemble iteration, post_iter.sh combines the log files from all of that
iteration’s segments into a single tar file.
west.h5¶
This file stores the simulation output in an hdf5 database. This includes the relationships between successive walkers, bin weights, progress coordinates, and auxiliary data.
west.log¶
This file contains a brief log of simulation progress. As WESTPA runs, it outputs information such as the current iteration number, the number of populated bins, and the time needed for each iteration in this log. This is also where errors are output.
Since only 10 iterations have been run, we do not yet have enough data to
analyze. Edit west.cfg and change max_total_iterations to 100. Extend
using the command:
./run.sh
The simulation will automatically continue from where it left off.
Computing the association rate¶
WESTPA includes several tools for analysis located in $WEST_ROOT/bin. In
init.sh we specified the bin containing an Na+/Cl-
distance of 1.8 Å as the bound state, and that containing a distance of 16.9 Å
as the unbound state. Using w_fluxanl, we can calculate the flux into these
target states, and from that calculate the association rate of Na+/Cl-. w_fluxanl may be run with the following commands:
source env.sh
$WEST_ROOT/bin/w_fluxanl
The script will output the flux into the target states including confidence intervals calculated using the block bootstrap method:
Calculating mean flux and confidence intervals for iterations [1,101)
target 'unbound':
correlation length = a tau
mean flux and CI = b (c, d) tau^(-1)
target 'bound':
correlation length = w tau
mean flux and CI = x (y, z) tau^(-1)
More information on how to use w_fluxanl can be viewed using the --help
flag. w_fluxanl also stores this information in an hdf5 file,
fluxanl.h5. Using the python libraries h5py and pylab, we can
visualize this data. Open a python interpreter and run the following commands:
import h5py, numpy, pylab
fluxanl = h5py.File('fluxanl.h5')
flux = numpy.zeros(100)
first_binding = 100 - fluxanl['target_flux']['target_1']['flux'].shape[0]
flux[first_binding:] = numpy.array(fluxanl['target_flux']['target_1']['flux'])
pylab.plot(flux)
pylab.xlabel("Iteration")
pylab.ylabel("Instantaneous Flux $(\\frac{1}{\\tau})$")
pylab.show()
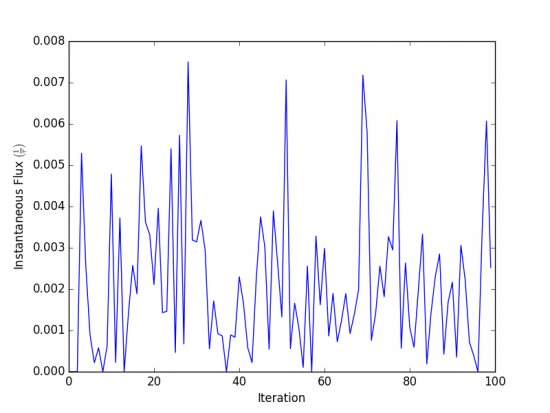
The x-axis represents the iteration number, and the y-axis the flux into the
bound state in units of τ-1 during that iteration. In the above
simulation, the first transition to the unbound state occurred in iteration 2,
and the first transition to the bound state occurred in iteration 3. The
instantaneous flux is noisy and difficult to interpret, and it is clearer to
view the time evolution of the flux. Run w_fluxanl again, this time with
the --evol flag:
$WEST_ROOT/bin/w_fluxanl --evol
We may plot the time evolution of flux using the following commands at a python interpreter:
import h5py, numpy, pylab
fluxanl = h5py.File('fluxanl.h5')
mean_flux = numpy.zeros(100)
ci_ub = numpy.zeros(100)
ci_lb = numpy.zeros(100)
first_binding = 100 - fluxanl['target_flux']['target_1']['flux_evolution']['expected'].shape[0]
mean_flux[first_binding:] = numpy.array(fluxanl['target_flux']['target_1']['flux_evolution']['expected'])
ci_lb[first_binding:] = numpy.array(fluxanl['target_flux']['target_1']['flux_evolution']['ci_lbound'])
ci_ub[first_binding:] = numpy.array(fluxanl['target_flux']['target_1']['flux_evolution']['ci_ubound'])
pylab.plot(mean_flux, 'b', ci_lb, 'g', ci_ub, 'r')
pylab.xlabel("Iteration")
pylab.ylabel("Mean Flux $(\\frac{1}{\\tau})$")
pylab.show()
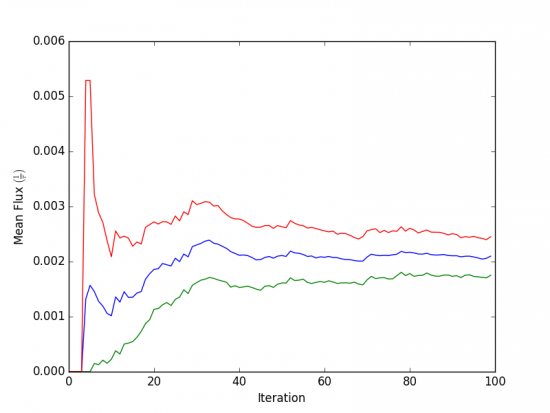
We can see that the flux has plateaued, indicating that the simulation has reached steady-state conditions. When calculating the rate, we discard the portion of data during which the system is equilibrating, using only portion over which the rates are steady and converging. We may calculate the rate using only the last 50 iterations:
$WEST_ROOT/bin/w_fluxanl --first-iter 50
Calculating mean flux and confidence intervals for iterations [50,101)
target 'unbound':
correlation length = 0 tau
mean flux and CI = 1.149779e-01 (1.094747e-01,1.209451e-01) tau^(-1)
target 'bound':
correlation length = 0 tau
mean flux and CI = 3.054023e-03 (2.378604e-03,3.863012e-03) tau^(-1)
Your output should be within an order of magnitude. Since τ for our simulation
was 0.5 ps, in order to determine the association rate in units of ps-1, the flux should be multiplied by 2, giving an association rate of
6.1 x 10-3 ps-1 with a 95% CI of 4.7 x10-3 to 7.7
x10-3. In order to obtain a more precise association rate, we would
need to run more iterations of the simulation, which may be done by editing
west.cfg.
Visualizing a selected pathway¶
Westpa includes the tools w_succ and w_trace to make concatenating
the segments for one of your completed pathways straightforward. Both
w_succ and w_trace are located in $WEST_ROOT/bin.
First use w_succ by entering into the command line from your simulation
root directory:
source env.sh
$WEST_ROOT/bin/w_succ
w_succ will output a list of every completed pathway, listed by its
iteration and segment ids. The target state each pathway has reached may be
determined from the final value of the progress coordinate. Pick any set of
completed iteration and segment ids and use them with the w_trace tool.
For example, if iteration 17 segment 2 is a completed pathway, run:
$WEST_ROOT/bin/w_trace 17:2
w_trace will output a text file named traj_17_2_trace.txt listing the
iteration and segment ids for the chain of continuing segments leading up to
the successful completion of your simulation. This file includes the iteration,
seg_id, weight, wallclock time, CPU time, and final progress coordinate value
for each segment comprising the trajectory. The first line, listed as iteration
0, includes the initial state ID. The same information is stored in hdf5 format
in the outfile trajs.h5.
By combining the information in this file with the coordinates stored in
west.h5, we can generate a complete trajectory viewable using Visual
Molecular Dynamics using the script
cat_trajectory.py, included in the westpa_scripts subfolder:
import h5py, numpy, sys
infile = numpy.loadtxt(sys.argv[1], usecols = (0, 1))
west = h5py.File('west.h5')
coords = []
for iteration, seg_id in infile[1:]:
iter_key = "iter_{0:08d}".format(int(iteration))
SOD = west['iterations'][iter_key]['auxdata']['coord'][seg_id,1:,0,:]
CLA = west['iterations'][iter_key]['auxdata']['coord'][seg_id,1:,1,:]
coords += [numpy.column_stack((SOD, CLA))]
with open(sys.argv[1][:-4] + ".xyz", 'w') as outfile:
for i, frame in enumerate(numpy.concatenate(coords)):
outfile.write("2\n")
outfile.write("{0}\n".format(i))
outfile.write("SOD {0:9.5f} {1:9.5f} {2:9.5f}\n".format(
float(frame[0]), float(frame[1]), float(frame[2])))
outfile.write("CLA {0:9.5f} {1:9.5f} {2:9.5f}\n".format(
float(frame[3]), float(frame[4]), float(frame[5])))
This script takes w_trace output as a command line argument, loads the
iteration and segment IDs, loads the coordinates for each segment from
west.h5`, and saves the results into an xyz file viewable using VMD.
Useful links¶
Useful hints¶
- Make sure your paths are set correctly in
env.sh. - If the simulation doesn’t stop properly with CTRL+C , use CTRL+Z.
- Another method to stop the simulation relatively cleanly is to rename
runseg.sh; WESTPA will shut the simulation down and prevent the hdf5 file from becoming corrupted. Some extra steps may be necessary to ensure that the analysis scripts can be run successfully.
